Simulating Glycan Shielding#
Proteins that are glycosylated have parts of their surface covered by the attached glycan chains. This makes them inaccessible for certain interactions such as with antibodies. To ascertain the “shielding effect” of glycans on a protein surface one can use molecular dynamics to obtain high quality conformers, albeit at a high computational cost.
Glycosylator offers a simple quick-simulation tool to sample glycan conformations on a glycosylated scaffold without the need for extensive computational resources. To that end Glycosylator provides the GlycoShield class and the top-level quickshield function. These will generate a dataframe containing an “exposure score” for each residue in the scaffold, which is a measure of how much a residue is “covered” by glycan chains and thus not exposed to the environment. Lower values indicate
higher coverage by glycans and vice versa.
[1]:
import glycosylator as gl
gl.visual.set_backend("py3dmol")
Using quickshield#
The most convenient way to simulate shielding is the quickshield function. It accepts a glycosylated Scaffold object as input and returns a GlycoShield which contains the data we are interested in. Let’s see how:
[2]:
# first load a scaffold
protein = gl.Protein.load("./files/protein_optimized.pkl")
for i, glycan in enumerate(protein.glycans):
glycan.id = f"glycan-{i}"
protein.show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
[5]:
# now run the quickshield
shield = gl.quickshield(protein)
|████████████████████████████████████████| 156/156 [100%] in 3:01.7 (0.86/s)
We can now inspect the exposure data by plotting it using plotly or matplotlib like so:
[11]:
shield.exposure.plotly().show()
/Users/noahhk/anaconda3/envs/glyco2/lib/python3.11/site-packages/plotly/express/_core.py:1985: FutureWarning:
When grouping with a length-1 list-like, you will need to pass a length-1 tuple to get_group in a future version of pandas. Pass `(name,)` instead of `name` to silence this warning.
Data type cannot be displayed: application/vnd.plotly.v1+json
We can actually visualize the “exposure” of the scaffold residues also as a heat map in the Py3DMol visualization like so:
[12]:
# show the results
shield.py3dmol().show()
You appear to be running in JupyterLab (or JavaScript failed to load for some other reason). You need to install the 3dmol extension:
jupyter labextension install jupyterlab_3dmol
And to obtain the actual data we can use the df attribute:
[13]:
shield.exposure.df
[13]:
| chain | resname | serial | exposure | |
|---|---|---|---|---|
| 0 | A | LEU | 401 | 249.983373 |
| 1 | A | GLU | 34 | 333.743532 |
| 2 | A | ARG | 153 | 134.149625 |
| 3 | A | ALA | 278 | 167.025529 |
| 4 | B | LEU | 495 | 177.947479 |
| ... | ... | ... | ... | ... |
| 573 | A | ILE | 400 | 270.514356 |
| 574 | A | TYR | 152 | 80.195666 |
| 575 | A | TYR | 277 | 208.773678 |
| 576 | A | THR | 33 | 324.323760 |
| 577 | B | VAL | 494 | 241.743316 |
578 rows × 4 columns
Using the GlycoShield directly#
The quickshield function is, well, quick… but it lacks some customizability. Let’s say we want to sample more edges for each glycan (the default is 3 edges per simulation) we will need to use the GlycoShield class directly. We can set it up by simply passing the scaffold to it as only argument and then use its simulate method to perform the shielding simulation like so:
[4]:
# setup the shield simulator
shield = gl.GlycoShield(protein)
# now run the simulation
out = shield.simulate(angle_step=50, repeats=3, edge_samples=5, save_conformations_to="./files/glycoshield", visualize_conformations=False)
|████████████████████████████████████████| 52/52 [100%] in 1:42.1 (0.51/s)
When providing visualize_conformations=True then out will be a tuple of the dataframe and a Py3DmolViewer. The visuals of the sampled (and accepted; ones with clashes are ignored) conformations are also stored by the GlycoShield so when we call py3dmol we can create a “full” view with the heatmap and conformations using:
shield.py3dmol().show()
Keep in mind though that depending on the size of the system this can be very, very, very….. memory intensive and thus slow down the code. It is usually better (and more likely useful anyway) to save the conformations into PDB files and then visualize them externally.
Anyways, this is what the glycoshield looks like when we assemble the exported PDB files:
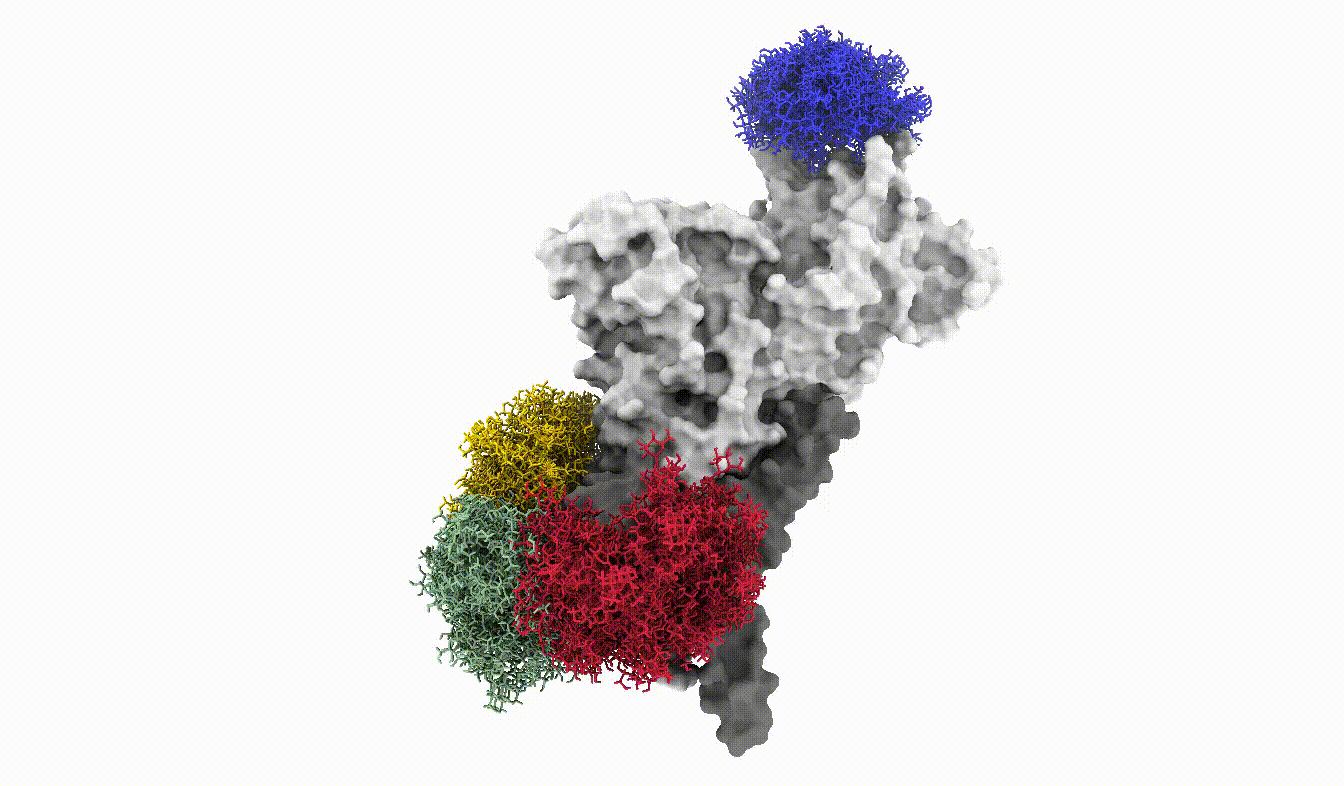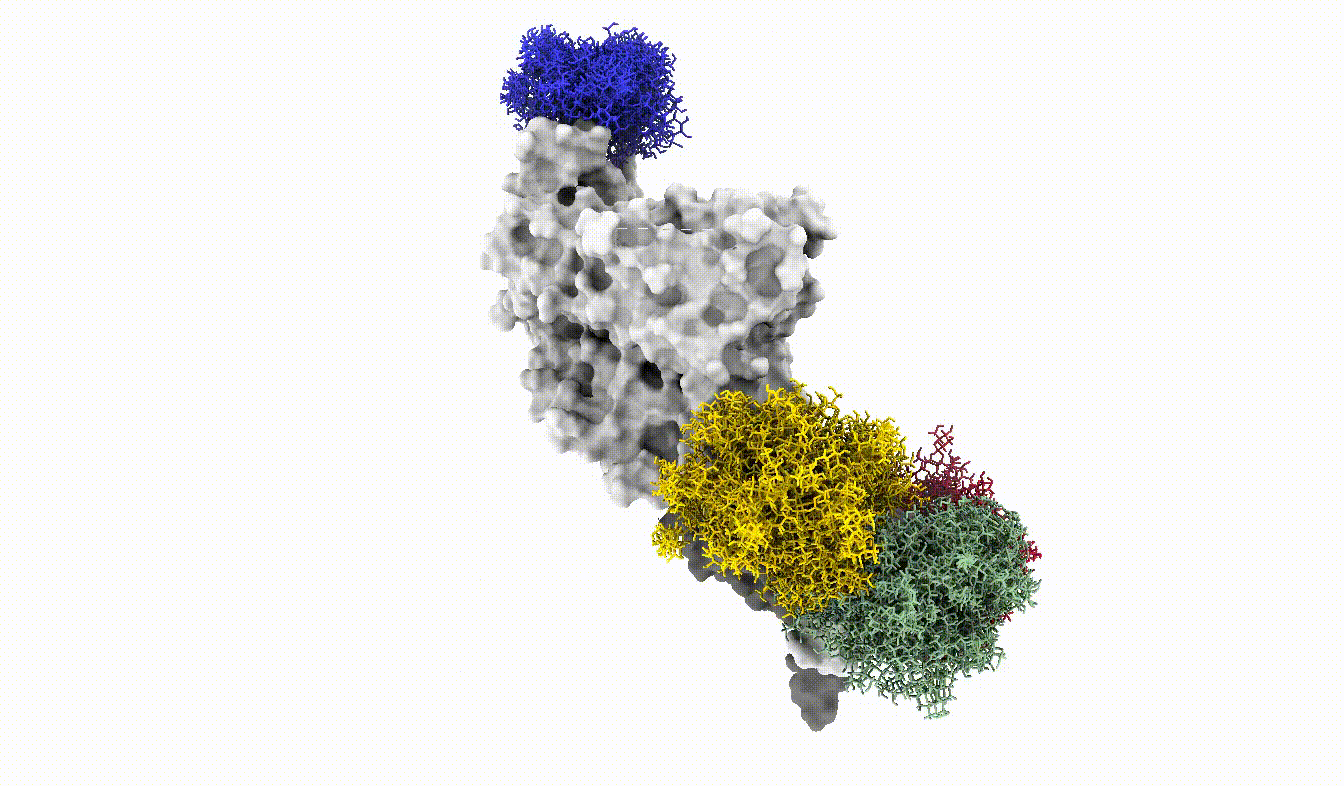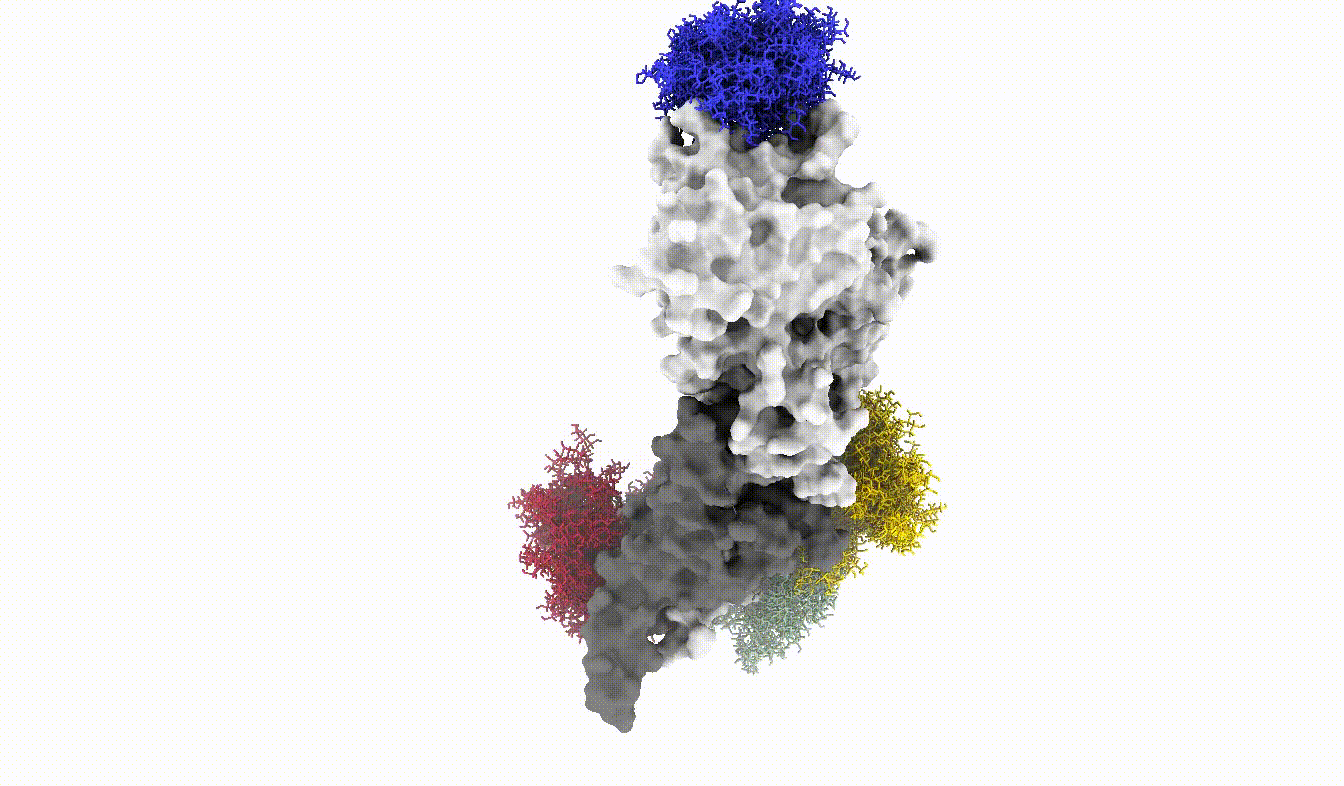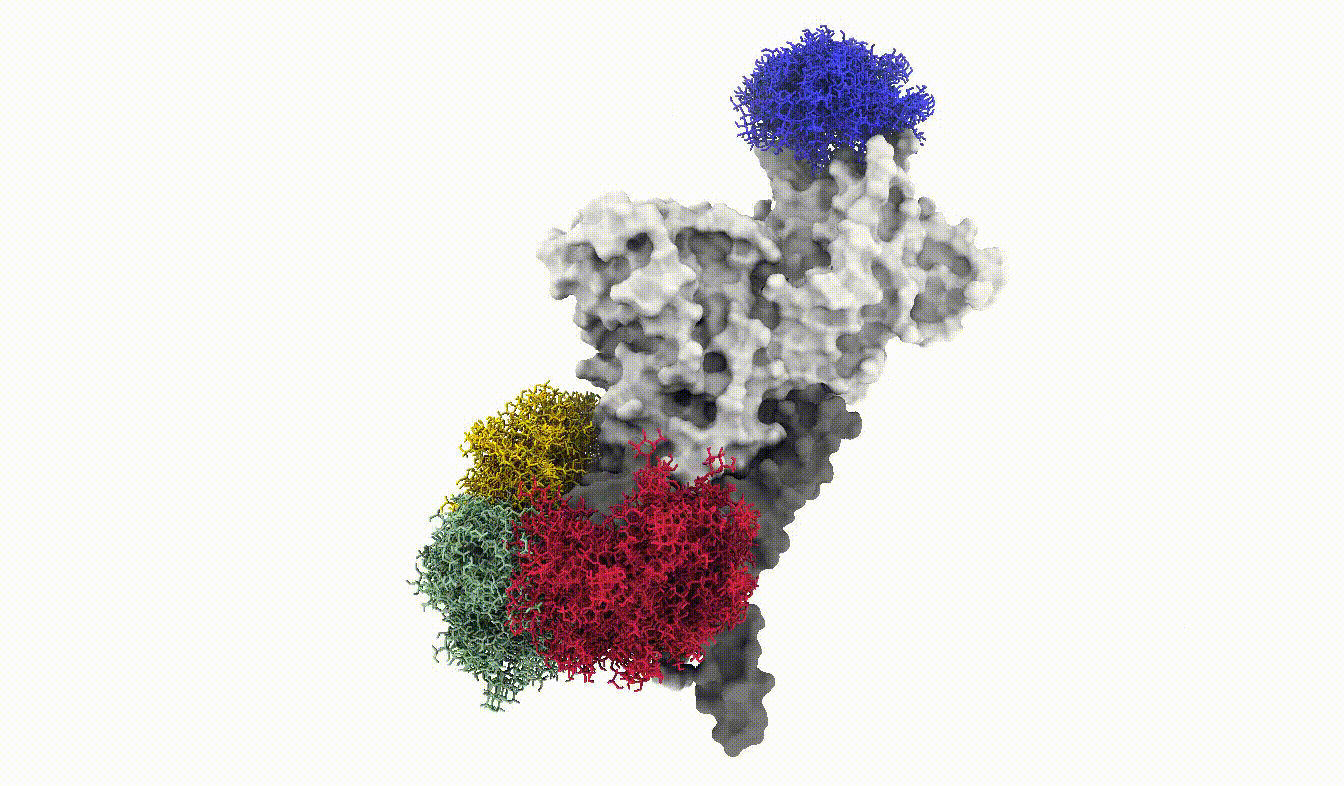
And with that we have reached the end of this tutorial. We have seen how we can use the GlycoShield class to perform sampling of glycan conformations on a scaffold such as a protein and obtain data on how covered or exposed certain residues are. Thanks for checking out this tutorial and good luck in your project using Glycosylator!